Below are the details and download links for other antibody-related tools produced by OPIG.
All the tools listed below are not actively maintained and with target dates to be deprecated.
ABangle
The relative orientation between the variable domains (VH and VL) influences the topology of the antigen binding site. It is important to consider when building high-resolution models of antibodies.
The VH/VL orientation can be represented by six measures:

| HL | the torsion angle between H1 and L1 |
| HC1 | the bend angle between H1 and C |
| HC2 | the bend angle between H2 and C |
| LC1 | the bend angle between L1 and C |
| LC2 | the bend angle between L2 and C |
| dc | the length of C |
Last updated: 30 May 2022
To be out of maintenance: 30 May 2022
EpiPred
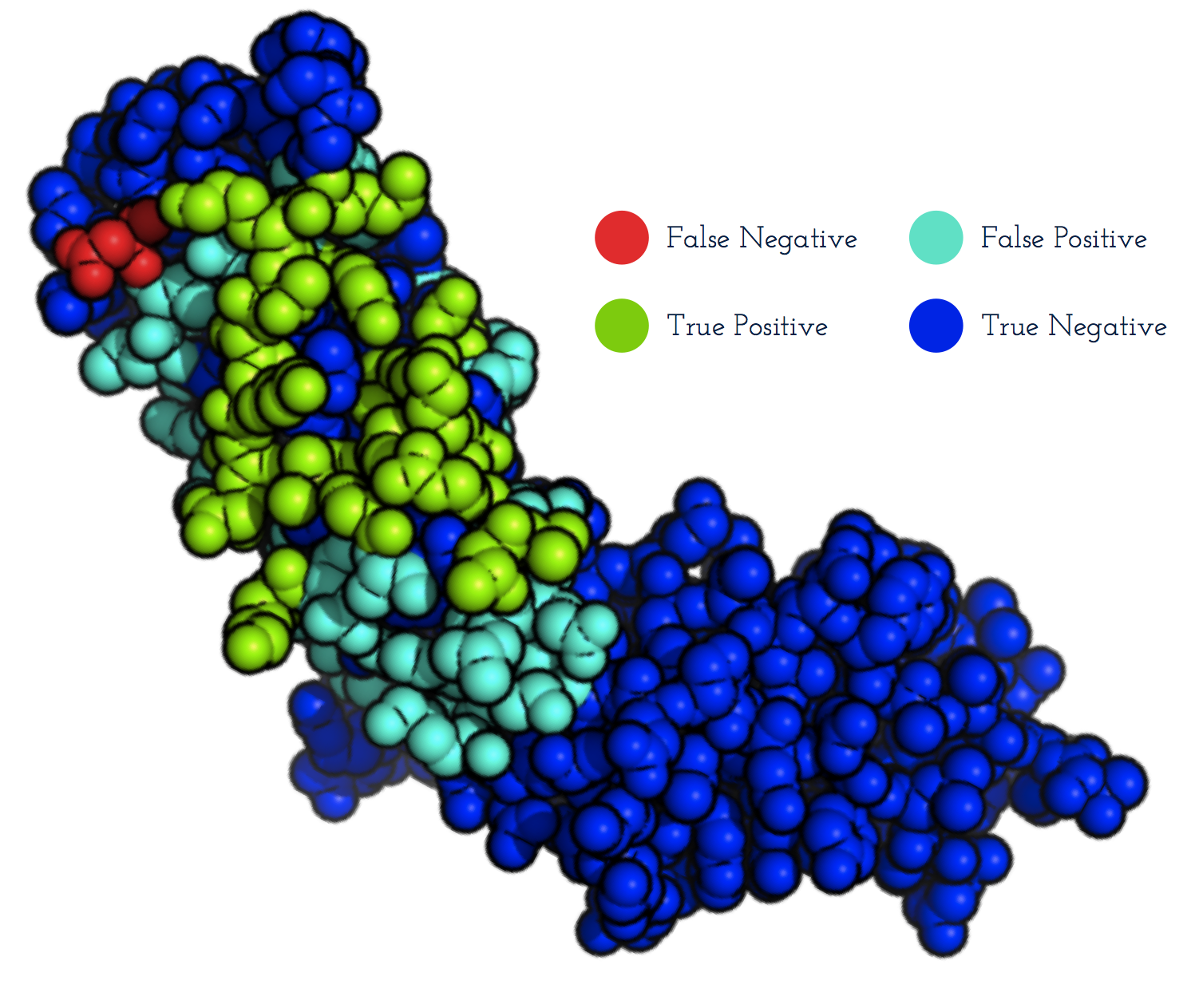
- EpiPred predicts structural epitopes specific to a given antibody.
- The method can be used with a homology model of the antibody as input.
- Patches on the antigen structure (right) are ranked according to how likely they are to be the epitope.
- The epitope predictions from EpiPred can be used to improve the performance of antibody-antigen docking.
Last updated: 30 May 2022
To be out of maintenance: 30 May 2022
Antibody i-Patch
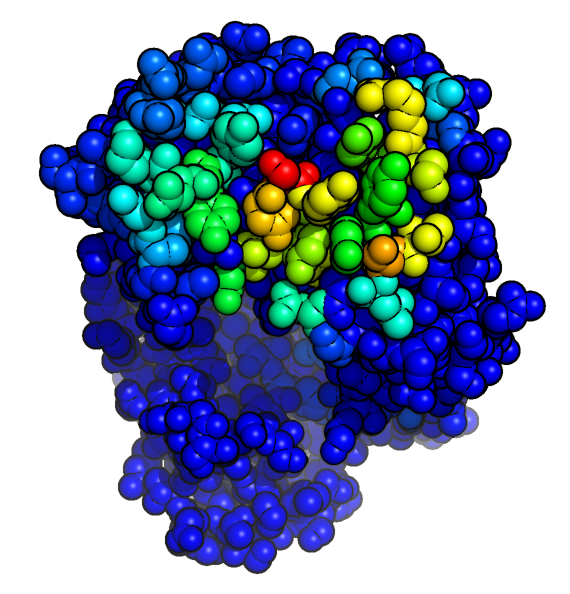
- Antibody i-Patch uses antibody-specific statistics to annotate residues with a score indicating how likely they are to be in contact with the supplied antigen.
- A higher score (warmer colours in the picture) indicates that the residue is more likely to form part of the paratope.
- The method can be used with a homology model of the antibody as input.
- Use the form below to submit an Antibody i-Patch job using the structures of an antibody-antigen pair.
Last updated: 30 May 2022
To be out of maintenance: 30 May 2022
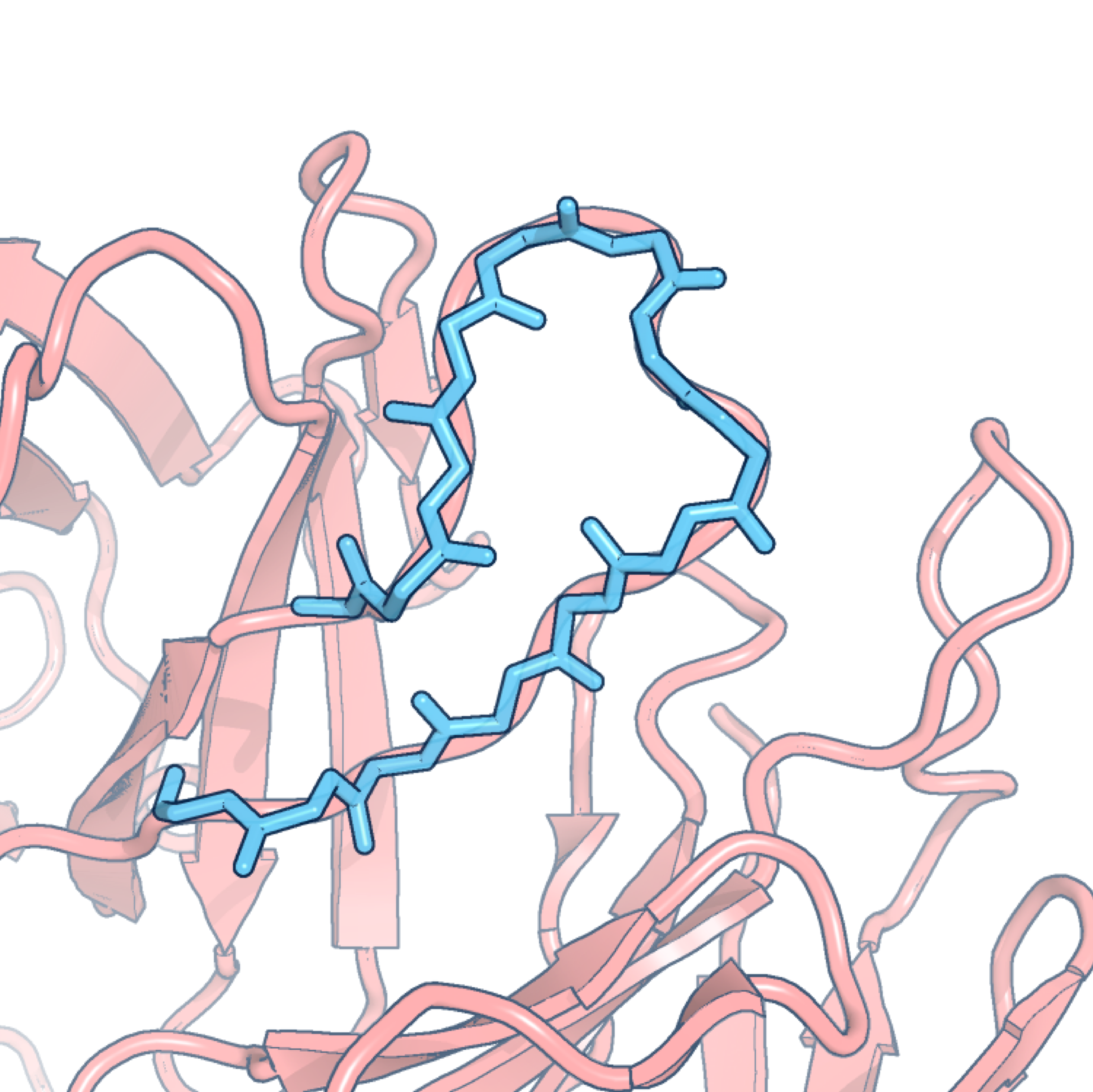
FREAD
FREAD is a database search loop structure prediction protocol. Its primary use is to fill in the gaps in incomplete 3D models of protein structures. Loops are generally located on the protein’s surface and their structures are known to be notoriously difficult to predict. The basic assumption of FREAD is that loops with similar sequences also have similar conformations, taking into account the spatial constraints introduced by their anchor residues (the residues on either side of the loop region).
FREAD predicts loop structures by selecting likely conformations from a database of experimentally-determined protein fragments. Fragments are selected according to the following criteria:
- Cα separations of anchor residues;
- Local sequence similarity measured by environment specific substitution tables (specified by dihedral angles);
- The absence of clashes if the fragment were to be inserted into the target structure;
- The RMSD between the anchors of the target and those of the fragment.
Last updated: 30 May 2022
To be out of maintenance: 30 May 2022
Sphinx
- Sphinx is a protein loop modelling algorithm that combines knowledge-based and ab initio approaches.
- Given a protein structure, location of the loop to be modelled and the sequence of that loop, it searches a database of fragments for sections of other proteins that are shorter than the target loop, but that may have some structural similarity. By using ab initio techniques, the loop conformations can be made to be the correct length.
- Once a set of conformations has been produced, a fast statistical potential is used to cull the set to only 500 structures, which are then scored using SOAP-Loop (Dong et al., 2013) to produce a ranking.
Last updated: 30 May 2022
To be out of maintenance: 30 May 2022