
N. B: This coordinate file can be downloaded directly from the PDB or generated using a prediction tool (e.g., ABodyBuilder2) from a VH-VL sequence.

Follow these steps:
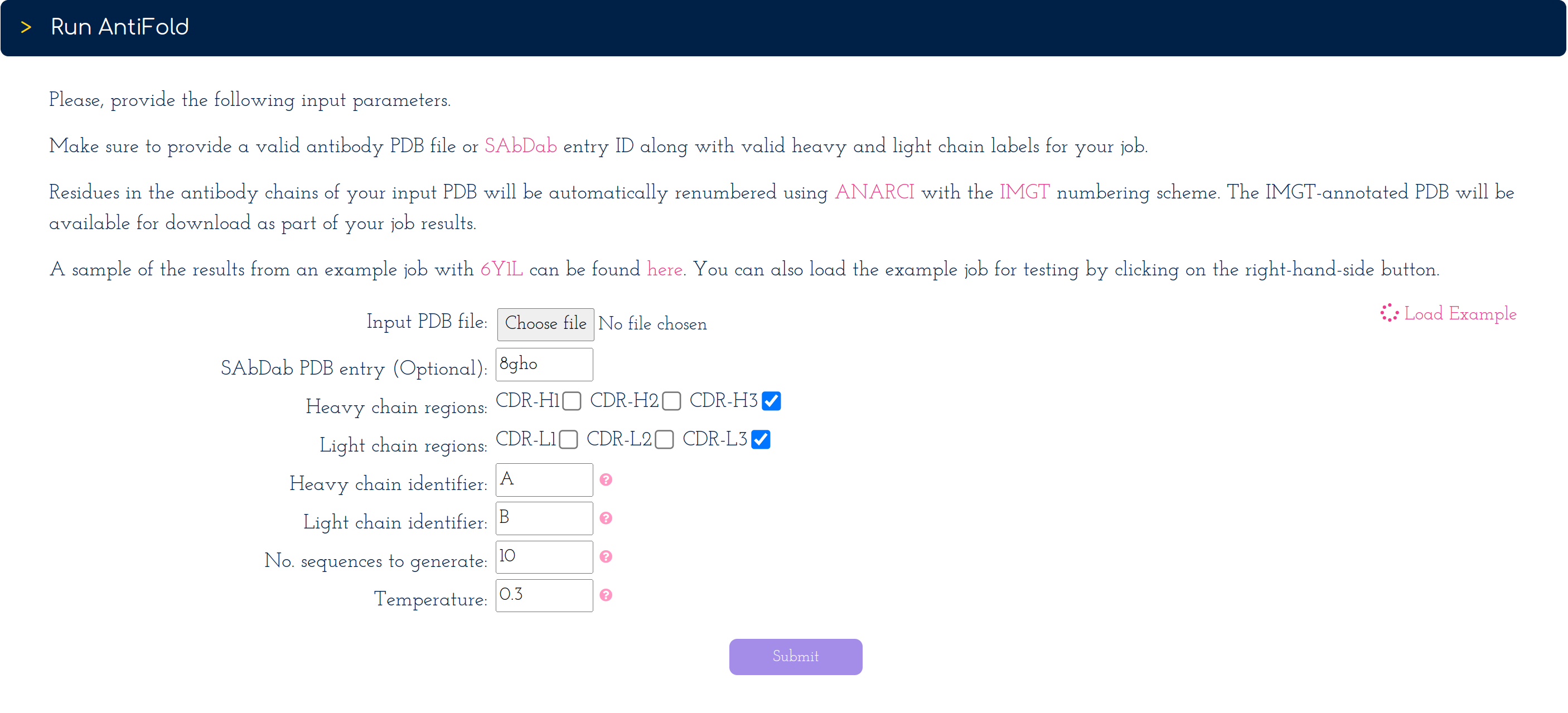
Follow these steps:
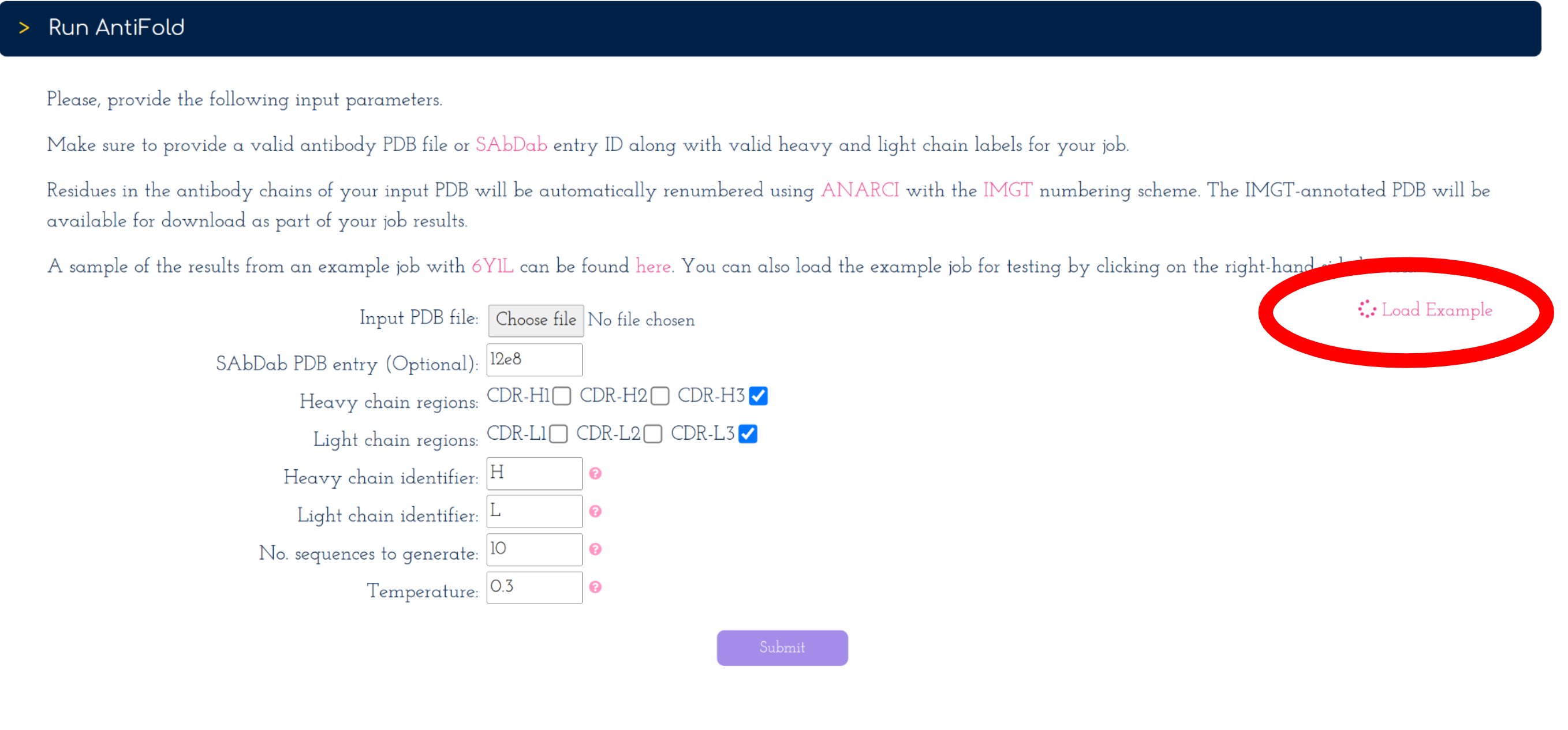
On the right-hand side of the submission form. You can click on the 'Load Example' button [See screenshot] to automatically load a PDB ID along with valid heavy and light chain identifiers.
As in scenarios 1 and 2, you can change all remaining parameters, if you wish.
After hitting the 'Submit' button, your job will be placed in a queue until finished.
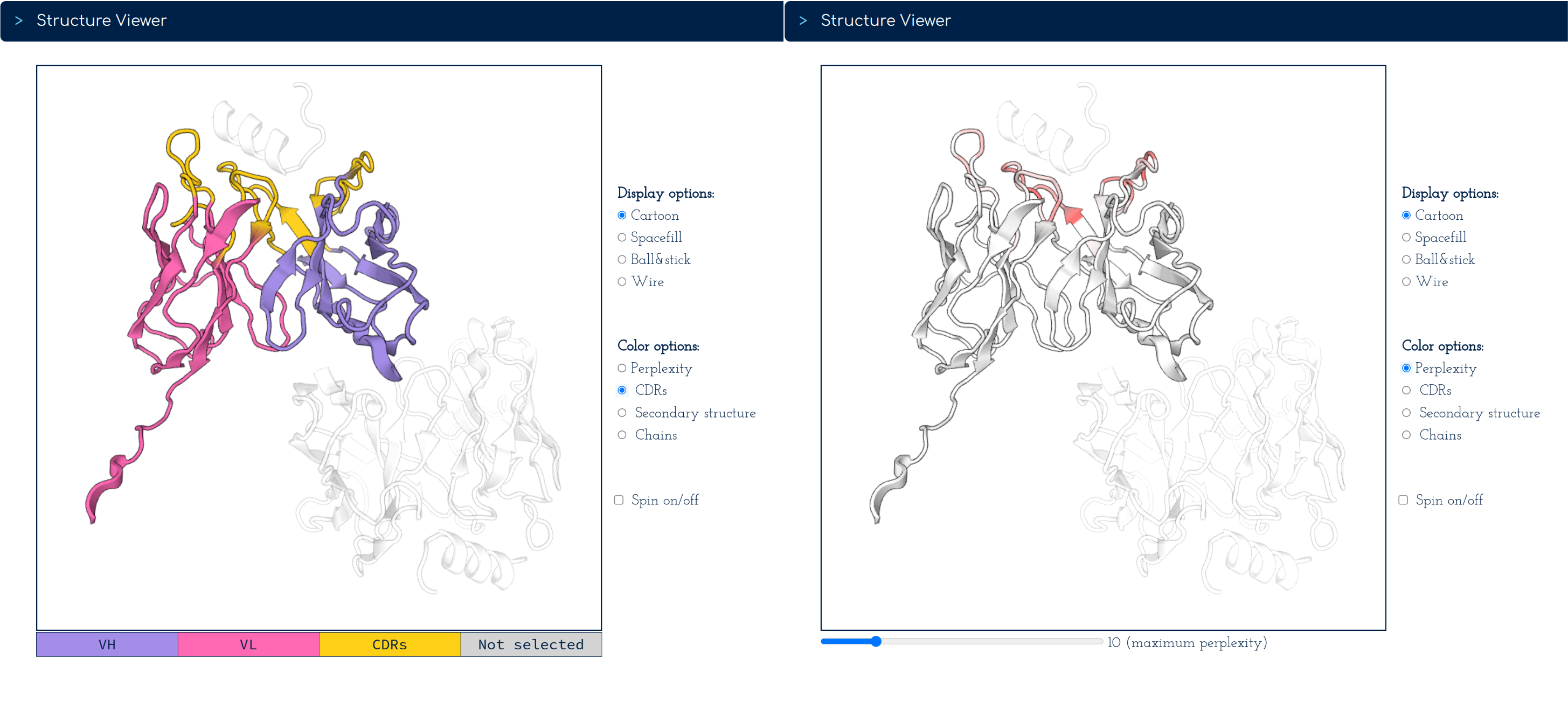
Perplexity at the residue level in the context of inverse folding is interpreted as the overall tolerance to mutations without altering the backbone structure.
The closer to perplexity equal one (1) a certain residue is, the less tolerant that residue position is to amino-acid variation. The opposite applies to high-perplexity positions.
The 'Structure Viewer' allows visualising AntiFold perplexity values of the CDR loops by clicking on the 'Perplexity' button under 'Color Options' [Right-hand-side screenshot].
To explore the perplexity CDR values for your input structure sequence the slider bar at the bottom of the viewer allows manipulation of the upper limit of the perplexity-encoded colour scale.
For instance, you can reveal which residues in the CDRs have the highest tolerance to mutation if they remain high in perplexity as you move the slider up.

The original and variant sequences, labelled Seq N, are tabulated to highlight (in red) at which CDR loop positions point mutations fitting the original backbone are present.
CDR 1 to 3 loops are colour-coded and their IMGT numberings (obtained using ANARCI) are found in the top row of the table.
In the logo plots underneath the table, the inverse folding probabilities for all 20 essential amino acids are mapped to the height of their amino acid letter and its opacity, both ranging from 0 to 100%.
For example, for the heavy chain in the screenshot, at residue 105 in the CDR-3, Alanine (A) has a higher probability (~ 90%), in contrast to the original residue, Therionine (T).
Note that since we didn't select the CDR-1 and CDR-2 regions, all these residues are identical for the sequence variants Seq 1 to Seq 10 - as expected.
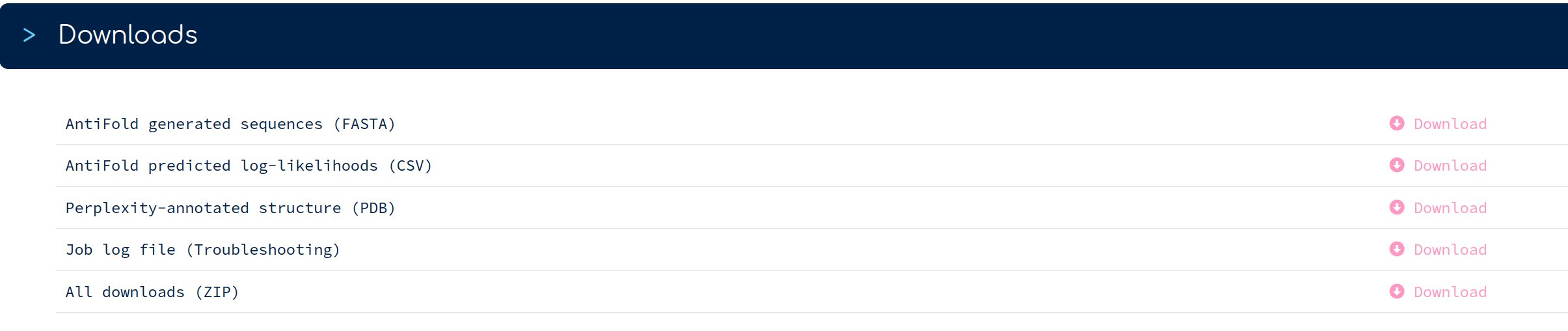
Here's a brief description of all file downloads, as listed:
Feel free to contact us at opig <~at~> stats.ox.ac.uk for any issues, or general enquiries about the AntiFold web app.
For specific issues about the AnitFold Python code, you can open an issue directly on the AntiFold GitHub repo.
Magnus Haraldson Høie & Alissa Hummer et al. “AntiFold:
Improved antibody structure-based design using inverse folding” 2024. [ link]
Download BibTex Reference
Header image credit: David Goodsell